Le talassemie sono un gruppo di malattie ereditarie caratterizzate da un difetto genetico di sintesi dell’emoglobina, la proteina contenuta nei globuli rossi del sangue. I difetti di una o più di queste catene causano le diverse sindromi talassemiche, in particolare le alfa-talassemie e le beta-talassemie.
Le Talassemie sono diffuse in tutto il mondo, l’Alfa talassemia si riscontra con maggiore frequenza in Africa, Asia meridionale, India, Paesi dell’est, Cina meridionale ed occasionalmente nei paesi del Mediterraneo.
A pagare maggiormente sono appunto i Paesi dell’area del Mediterraneo, ma anche il Medio Oriente e il Sudest asiatico (indiano, pakistano, ecc.).
In Africa ogni anno nascono 120 mila bambini affetti da varie forme di anemia. La forte preponderanza delle diverse varianti della malattia sul continente africano si riverbera anche in America, dove si contano almeno 50 mila pazienti, soprattutto tra la popolazione afroamericana (9%). Si stima che le persone affette da questa malattia siano circa 3 milioni con circa 50 mila nuovi casi ogni anno.
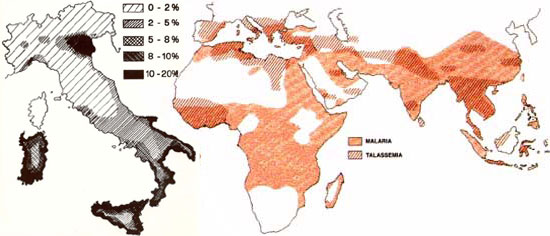
In Italia, è particolarmente diffusa in alcune regioni quali il Piemonte, la Calabria, la Sicilia, la Puglia e la Sardegna; i malati sono circa 9000 solo nel Delta Padano, nel sud, in Sicilia e in Sardegna.
In ogni caso nel mondo i portatori sani di anemia mediterranea sono oltre 300 milioni. Di questi, 93 milioni sono bambini.
Esistono tre maggiori forme di Beta-talassemie: Minor, Intermedia e la forma più grave, Talassemia Major o Morbo di Cooley, dove la produzione insufficiente di emoglobina può determinare gravi danni agli organi interni.
La Talassemia, o Anemia Mediterranea, o Morbo di Cooley, è un’anemia molto grave dovuta ad un difetto di formazione dell’emoglobina. Proprio per mezzo dell’emoglobina è possibile la formazione dei globuli rossi, i quali sono i principali vettori dell’ossigeno dai polmoni lungo tutto il corpo. In una persona affetta da talassemia i globuli rossi non possono essere prodotti normalmente a causa della mancata formazione dell’emoglobina. Per sopperire alla mancanza di emoglobina, i talassemici sono costretti a sottoporsi, fin dai primi mesi di vita, a continue trasfusioni di sangue ogni 20/30 giorni.
La conseguenza a questo intenso apporto di globuli rossi è un’alterazione a carico del cuore, del fegato, della milza, dello scheletro. Infatti le frequenti trasfusioni portano ad un sovraccarico di “ferro”, un metallo prezioso per il nostro organismo, ma se in quantità elevate incompatibile con la vita. L’accumulo eccesivo di ferro in tutti gli organi costringe i talassemici a specifiche terapie quotidiane “deferrizzanti” che diventano essenziali per mantenersi in buona salute; esse eliminano una parte di ferro in eccesso che altrimenti determinerebbe patologie gravi e precoci a tali organi, portando il soggetto alla morte.

La talassemia è una malattia ereditaria e per trasmetterla occorre essere microcitemici (portatori sani); ciò significa avere nel proprio codice genetico (DNA) una caratteristica particolare (tratto talassemico) che rende possibile la trasmissione della malattia.
L’arma più efficace per evitare che la talassemia continui a diffondersi è la PREVENZIONE.
Le molecole alfa e beta, costituenti dell’emoglobina, nella talassemia presentano un difetto in una delle due catene; il risultato prodotto è uno scarso livello di emoglobina nel sangue ed il colorito dei globuli e rosso pallido.
BIBLIOGRAFIA
Linee guida per il trattamento clinico della talassemia 2a edizione aggiornata (2008): guidelines_italianjan2010_2
Linee guida per la prevenzione ed il trattamento delle complicanze della talassemia (1999):
prev-cura-complicanze
La Talassemia (2007):
about-thalassaemia-2007-italian
Disordini genetici dell’emoglobina o emoglobinopatie – Beta Talassemia (2007):
booklet-a-thalassaemia-italian
Disordini genetici dell’emoglobina o emoglobinopatie – Alfa Talassemia (2007):
booklet-b-thalassaemia-italian
[:en]Le talassemie sono un gruppo di malattie ereditarie caratterizzate da un difetto genetico di sintesi dell’emoglobina, la proteina contenuta nei globuli rossi del sangue. I difetti di una o più di queste catene causano le diverse sindromi talassemiche, in particolare le alfa-talassemie e le beta-talassemie. Le Talassemie sono diffuse in tutto il mondo, l’Alfa talassemia si riscontra con maggiore frequenza in Africa, Asia meridionale, India, Paesi dell’est, Cina meridionale ed occasionalmente nei paesi del Mediterraneo.
A pagare maggiormente sono appunto i Paesi dell’area del Mediterraneo, ma anche il Medio Oriente e il Sudest asiatico (indiano, pakistano, ecc.). In Africa ogni anno nascono 120 mila bambini affetti da varie forme di anemia. La forte preponderanza delle diverse varianti della malattia sul continente africano si riverbera anche in America, dove si contano almeno 50 mila pazienti, soprattutto tra la popolazione afroamericana (9%). Si stima che le persone affette da questa malattia siano circa 3 milioni con circa 50 mila nuovi casi ogni anno.
In Italia, è particolarmente diffusa in alcune regioni quali il Piemonte, la Calabria, la Sicilia, la Puglia e la Sardegna; i malati sono circa 9000 solo nel Delta Padano, nel sud, in Sicilia e in Sardegna.
In ogni caso nel mondo i portatori sani di anemia mediterranea sono oltre 300 milioni. Di questi, 93 milioni sono bambini.
Esistono tre maggiori forme di Beta-talassemie: Minor, Intermedia e la forma più grave, Talassemia Major o Morbo di Cooley, dove la produzione insufficiente di emoglobina può determinare gravi danni agli organi interni.









